
Chemical synthesis is essential in the development of new molecules for medical applications, materials science and fine chemistry. This process, which involves planning chemical reactions to create desired target molecules, has traditionally relied on human expertise. Recent advances have turned to computational methods to improve the efficiency of retrosynthesis: working backwards from a target molecule to determine the series of reactions necessary to synthesize it. Leveraging modern computational techniques, researchers aim to solve long-standing bottlenecks in synthetic chemistry, making these processes faster and more precise.
One of the critical challenges in retrosynthesis is to accurately predict chemical reactions that are rare or less frequently encountered. These reactions, although rare, are vital for designing new chemical pathways. Traditional machine learning models often fail to predict these reactions due to underrepresentation in the training data. Furthermore, multi-step retrosynthesis planning errors can cascade, leading to invalid synthetic routes. This limitation hinders the ability to explore innovative and diverse avenues for chemical synthesis, particularly in cases requiring unusual reactions.
Existing computational methods for retrosynthesis have primarily focused on one-step models or rule-based expert systems. These methods rely on predefined rules or large training data sets, which limits their adaptability to new and unique reaction types. For example, some approaches use graph- or sequence-based models to predict the most likely transformations. While these methods have improved the precision of common reactions, they often require more flexibility to account for the complexities and nuances of rare chemical transformations, creating a gap in comprehensive retrosynthetic planning.
Researchers from Microsoft Research, Novartis Biomedical Research, and Jagiellonian University developed Chimera, a joint framework for retrosynthesis prediction. Chimera integrates results from multiple machine learning models with various inductive biases, combining their strengths through a learned classification mechanism. This approach leverages two recently developed state-of-the-art models: NeuralLoc, which focuses on molecule editing using graph neural networks, and R-SMILES 2, a de novo model employing a sequence-to-sequence Transformer architecture. By combining these models, Chimera improves both the accuracy and scalability of retrosynthetic predictions.
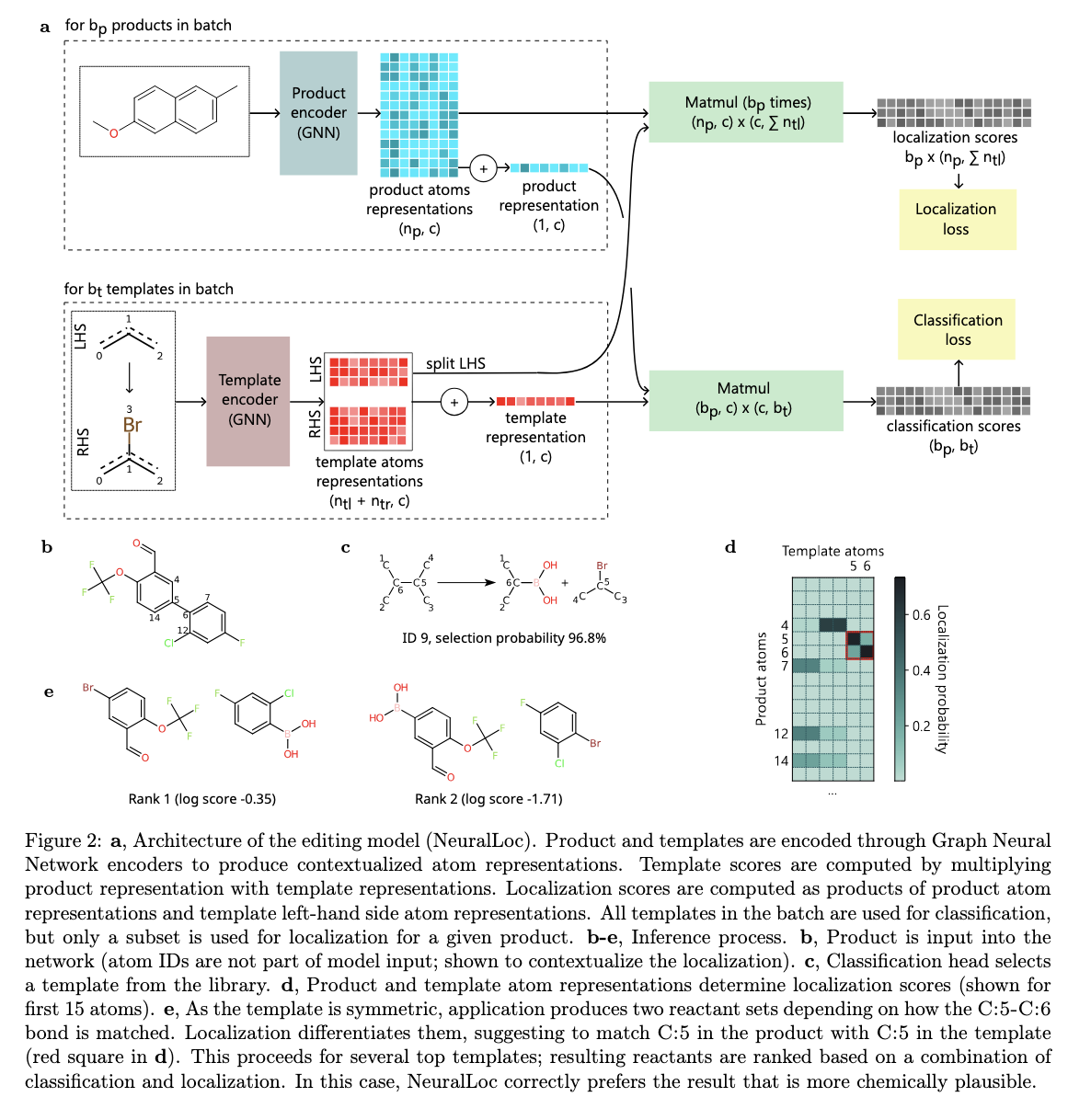
The methodology behind Chimera is based on combining the results of its constituent models through a ranking system that assigns scores based on model agreement and predictive confidence. NeuralLoc encodes molecular structures as graphs, allowing accurate prediction of reaction sites and templates. This method ensures that the predicted transformations closely align with known chemical rules while maintaining computational efficiency. Meanwhile, R-SMILES 2 uses advanced attention mechanisms, including group inquiry attention, to predict reaction pathways. The architecture of this model also incorporates improvements to the normalization and activation functions, ensuring superior gradient flow and inference speed. Chimera combines these predictions, using overlap-based scores to rank potential pathways. This integration ensures that the framework balances the strengths of editing-based and de novo approaches, allowing robust predictions even for complex and rare reactions.
Chimera's performance has been rigorously validated against publicly available datasets such as USPTO-50K and USPTO-FULL, as well as the proprietary Pistachio dataset. In USPTO-50K, Chimera achieved a 1.7% improvement in the accuracy of the top 10 predictions over previous state-of-the-art methods, demonstrating its ability to accurately predict both common and rare reactions. In USPTO-FULL, it further improved the accuracy of the top 10 by 1.6%. Extending the model to the Pistachio data set, which contains more than three times the USPTO-FULL data, showed that Chimera maintained high accuracy over a broader range of reactions. Comparisons of experts with organic chemists revealed that Chimera predictions were consistently preferred over individual models, confirming its effectiveness in practical applications.
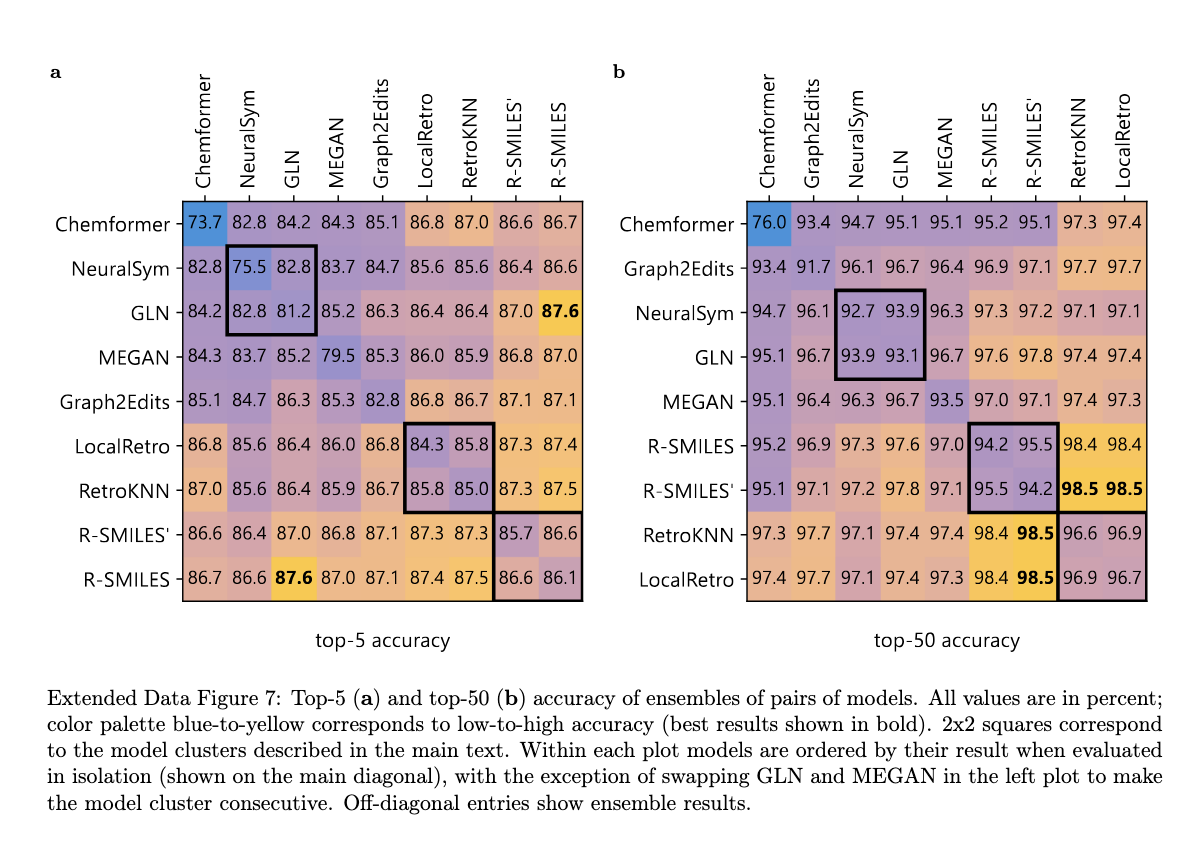
The framework was also tested on an internal Novartis data set of over 10,000 reactions to assess its robustness to distribution changes. In this zero-shot configuration, where no additional adjustments were made, Chimera demonstrated superior accuracy compared to its constituent models. This highlights its ability to generalize across data sets and predict viable synthetic routes even in real-world scenarios. Additionally, Chimera excelled in multi-step retrosynthesis tasks, achieving success rates close to 100% on benchmarks such as SimpRetro, significantly outperforming individual models. The framework's ability to find pathways for highly challenging molecules further underscores its potential to transform computational retrosynthesis.
Chimera represents a groundbreaking advance in retrosynthesis prediction by addressing the challenges of rare reaction prediction and multi-step planning. The framework demonstrates superior accuracy and scalability by integrating diverse models and employing a robust classification mechanism. With its ability to generalize across data sets and excel at complex retrosynthetic tasks, Chimera is poised to accelerate progress in chemical synthesis, paving the way for innovative approaches to molecular design.
Verify he Paper. All credit for this research goes to the researchers of this project. Also, don't forget to follow us on <a target="_blank" href="https://twitter.com/Marktechpost”>twitter and join our Telegram channel and LinkedIn Grabove. Don't forget to join our SubReddit over 60,000 ml.
Trending: LG ai Research launches EXAONE 3.5 – three frontier-level bilingual open-source ai models that deliver unmatched instruction following and broad context understanding for global leadership in generative ai excellence….
Nikhil is an internal consultant at Marktechpost. He is pursuing an integrated double degree in Materials at the Indian Institute of technology Kharagpur. Nikhil is an ai/ML enthusiast who is always researching applications in fields like biomaterials and biomedical science. With a strong background in materials science, he is exploring new advances and creating opportunities to contribute.
<script async src="//platform.twitter.com/widgets.js” charset=”utf-8″>






{kind=link}