 NEWSLETTER
NEWSLETTER
The growing urgency of innovative medicines in various medical fields, such as antibiotics, cancer treatments, autoimmune disorders, and antiviral therapies, underscores the need to increase research and development efforts. Drug discovery, a complex process involving the exploration of a vast chemical space, can benefit from computational methods and, more recently, deep learning. Deep learning, particularly generative ai, shows promise for efficiently screening extensive chemical libraries, predicting new bioactive molecules, and improving drug candidate development through learning and pattern recognition over time.
Researchers from the Faculty of Medicine, University of Porto, Porto, Portugal, Department of Community Medicine, Information and Decision in Health, Faculty of Medicine, University of Porto, Porto, Portugal, Center for Research in Health Technologies and Services (CINTESIS) , Porto, Portugal, Faculty of Health Sciences, Fernando Pessoa University, Porto, Portugal, SIGIL Scientific Enterprises, Dubai, United Arab Emirates and MedFacts Lda., Lisbon, Portugal have created WithHOWEVER. This deep learning model uses Wasserstein generative adversarial networks and graph convolutional networks. Their goal is to generate new quinoline scaffold molecules by working with intricate molecular graphics. The development process involved tuning hyperparameters and evaluating drug-like qualities such as pharmacokinetics, toxicity, and synthetic accessibility.
The study analyzes the urgent need for new and effective drugs in various classes, such as antibiotics, anticancer treatments, autoimmune disorders and antiviral treatments, due to emerging challenges in drug delivery, disease mechanisms and rapid rates of mutation. It highlights the potential of generative ai in drug discovery, including drug repurposing, optimization and de novo design, using techniques such as recursive neural networks, autoencoders, generative adversarial networks and reinforcement learning. The study emphasizes the importance of exploring the vast chemical space for drug discovery and the role of computational methods in guiding the process toward optimal targets.
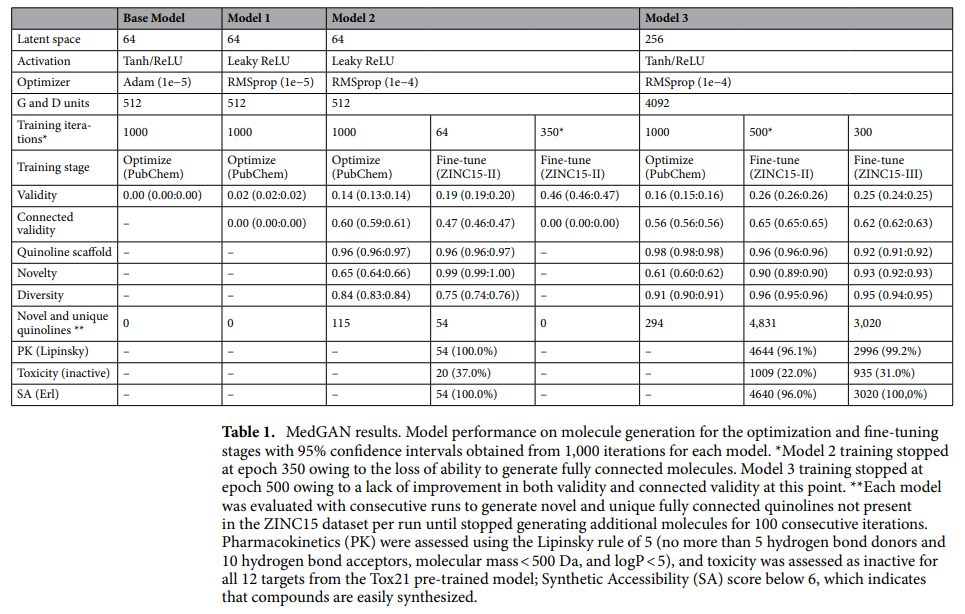
The study used the WGAN architecture to develop a new GAN model to create quinoline-like molecules. The goal was to improve and optimize the model output by emphasizing the learning of particular key patterns, such as the molecular structure inherent to the quinoline structure. The model was fitted using an optimized GAN approach, where three different models (models 1, 2 and 3) were trained and evaluated based on their ability to generate valid chemical structures. Models 2 and 3 showed a marked improvement over the base model, achieving higher scores in the development of valid chemical structures. These models were selected for further fitting using a larger data set of quinoline molecules.
The study also divided the ZINC15 data set into three subsets based on complexity, which were used sequentially to refine the training. The subsets included quinoline molecules of different sizes and constitutions, allowing for a more tailored approach to generating molecules with superior chemical properties.
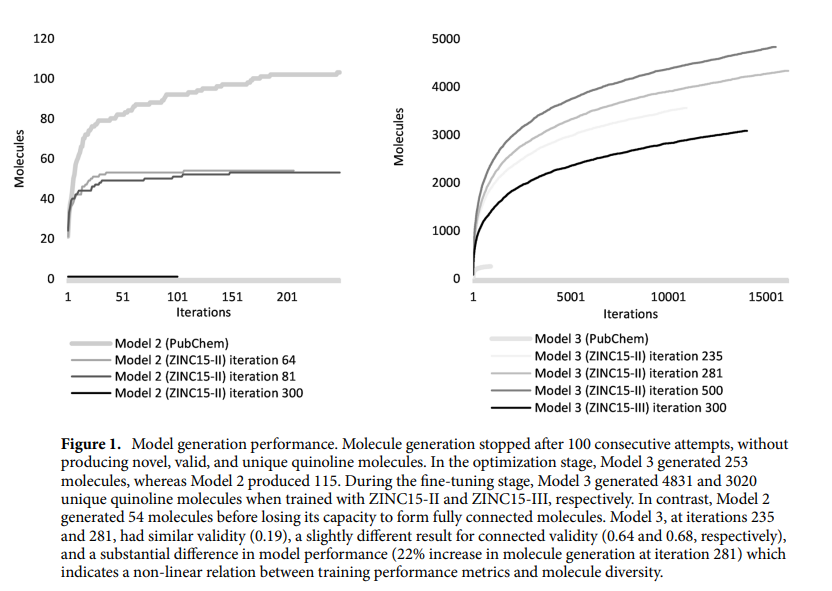
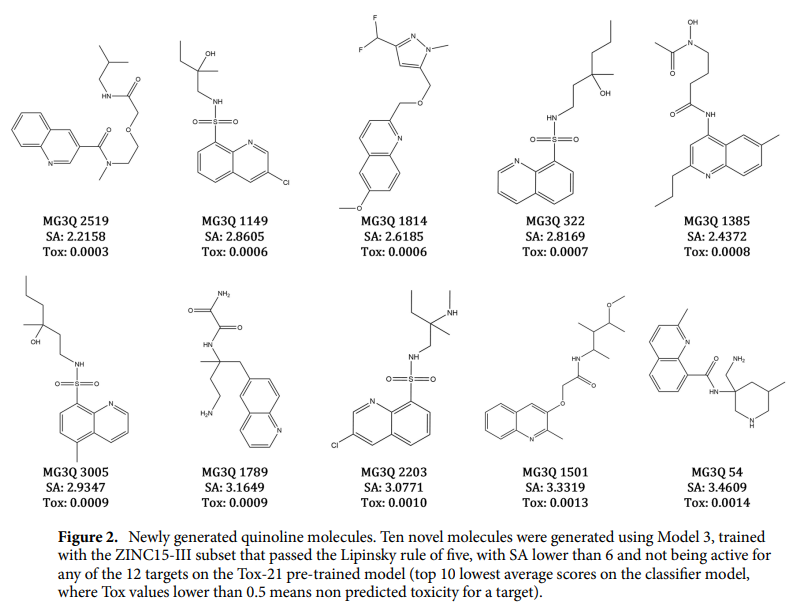
The MedGAN model has been optimized to create quinoline scaffold molecules for drug discovery and has achieved impressive results. The best model developed 25% valid and 62% fully connected molecules, of which 92% were quinolines and 93% were unique. It retained important properties such as chirality, atomic charge, and favorable drug-like attributes. It successfully generated 4831 unique and fully connected quinoline molecules that were not present in the original training data set. These generated molecules adhere to Lipinski's rule of 5, which indicates their potential bioavailability and synthetic accessibility.
In conclusion, the study presents MedGAN, a GCN-optimized GAN for molecule design. The generated molecules retained important drug-like properties, including chirality, atomic charge, and favorable pharmacokinetics. The model demonstrated the potential to create new molecular structures and improve deep learning applications in computational drug design. The study highlights the impact of several factors, such as activation functions, optimizers, learning rates, molecule size, and scaffold structure, on the performance of generative models. MedGAN offers a promising approach to rapidly access and explore chemical libraries, discovering new patterns and interconnections for drug discovery.
Review the Paper and GitHub. All credit for this research goes to the researchers of this project. Also, don't forget to follow us on Twitter. Join our 36k+ ML SubReddit, 41k+ Facebook community, Discord channeland LinkedIn Grabove.
If you like our work, you will love our Newsletter..
Don't forget to join our Telegram channel
![]()
Sana Hassan, a consulting intern at Marktechpost and a dual degree student at IIT Madras, is passionate about applying technology and artificial intelligence to address real-world challenges. With a strong interest in solving practical problems, she brings a new perspective to the intersection of ai and real-life solutions.
<!– ai CONTENT END 2 –>






{kind=link}